Painful swellings that appear suddenly (1). Maybe together with nausea, vomiting, abdominal pain (2)… It comes naturally to ask: is it an allergy attack? It’s not said. It could be hereditary angioedema, a rare disease still the subject of studies and research, which is often diagnosed 10 years late (3).
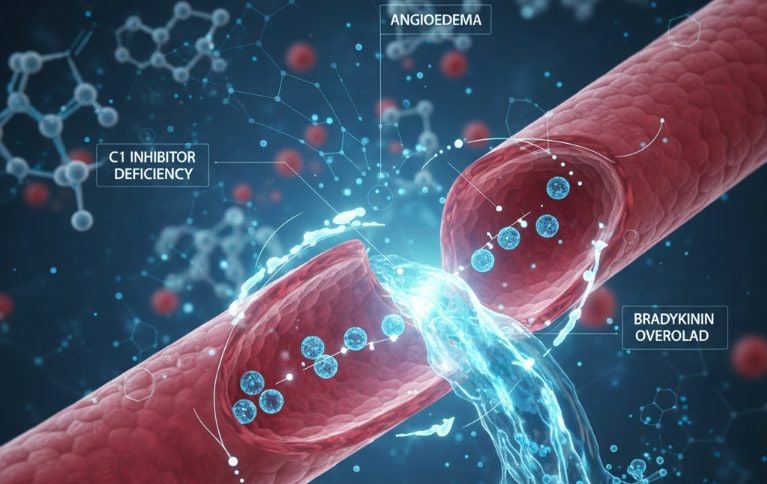
The cause of this pathology is a genetic mutation (4) that prevents the body from correctly producing a protein called C1 inhibitor (4). This creates a cascade effect: bradykinin, a substance that dilates blood vessels, causes water to leak into one part of the body, resulting in swelling called angioedema (4). Hereditary angioedema can be fatal if not recognized and treated early (5). For those who suffer from it, the day becomes a real gamble: will the attack come today or not? And when it arrives, will I be able to talk, work, drive, fly?
Living with an illness from which you cannot recover is difficult, but it can be kept under control with targeted therapies.
What is hereditary angioedema and how does it manifest itself
Hereditary angioedema is one rare genetic disease which usually manifests itself with sudden attacks of edema during childhood or adolescence, but which can take many years before being recognized. (6)
The pathology is transmitted with autosomal dominant mode (7): this means that it is enough to develop it inherit a single mutated copy of the gene involved by one of the parents. However, about a quarter of people who suffer from it have a spontaneous mutation, which has manifested itself de novo (8), i.e. without there being a family history of the disease.
This mutation in DNA causes the body to not produce enough of a protein called C1 inhibitor (4) (fundamental for the balance of different physiological systems), or that produces it, but it doesn’t work properly. When the C1 inhibitor is missing or not working, the body produces too much bradykinin, a substance that makes blood vessels more “open”, by releasing liquids and causing sudden swelling (angioedema attacks) (4).
The disease is characterized by edema recurrent that can affect the skin, mucous membranes or internal organs. Untreated attacks typically last 48 to 96 hours. Bloating is often associated with pain, nausea, vomiting, diarrhea and swelling of the airways, the latter potentially lethal: if not treated in time, the risk of death from airway obstruction is significant. (9)
Given the symptoms, at first doctors might lean towards severe allergic crises, but standard anti-allergic therapeutic approaches may prove to be totally ineffective. (10) These attacks can be triggered by small physical traumas, stress, infections and hormonal changes, but most of the time they occur without particular triggers. (11)
In any case, the unpredictability of these attacks generates constant anticipatory anxiety in people suffering from angioedema, which limits the freedom to plan activities, trips and appointments. In many of them, a sense of fragility also develops and social relationships are compromised, due to the difficulty of making and maintaining programs. (12)
New perspectives on life for those suffering from hereditary angioedema
In recent years, therapeutic innovation has managed to greatly improve the lives of these patients, both in terms of acute attacks and prophylaxis. In fact, thanks to increasingly personalized therapeutic paths and reference centers present throughout the country, it is possible not only to uncover cases that have not yet been diagnosed, but also and above all to better manage the pathology, also thanks to targeted therapies that prevent acute attacks.
In Italy, the initiative “Angioedema Real Life. Logbook towards Ithaca” promoted by Takeda Italia (with the patronage of the Voluntary Association for Hereditary Angioedema and other rare forms of angioedema, ITACA, SIAAIC, SIAIP and AAIITO). The heart of the project is an exciting video diary in six episodes that explores first-hand the lives of people living with this disease and the crucial role of the ITACA Centers in the diagnosis and treatment process. A journey to be observed with great attention, to understand how angioedema affects the life, affections and projects of those affected by it.
Bibliography
– Wong JCY, Cheong N, Lau CS, Li PH. Prevalence and impact of misdiagnosed drug allergy labels among patients with hereditary angioedema. Front Allergy. 2022 Aug 16;3:953117. doi: 10.3389/falgy.2022.953117. PMID: 36051898; PMCID: PMC9426770.
1. Banerji A. The burden of illness in patients with hereditary angioedema. Ann Allergy Asthma Immunol. 2013;111(5):329-336.
2. Zuraw BL. Hereditary angioedema. N Engl J Med. 2008;359(10):1027-1036.
3. Zanichelli A, Longhurst HJ, Maurer M, Bouillet L, Aberer W, Fabien V, Andresen I, Caballero T; IOS Study Group. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy Asthma Immunol. 2016 Oct;117(4):394-398. doi: 10.1016/j.anai.2016.08.014. PMID: 27742086.
4. HEREDITARY ANGIOEDEMA due to C1 inhibitor defect edited by Marco Cicardi, Andrea Zanichelli L. Sacco Department of Biomedical and Clinical Sciences, University of Milan, L. Sacco Hospital, Milan
5. Gayatri Patel, Jacqueline A Pongracic. Hereditary and acquired angioedema Allergy Asthma Proc. 2019 Nov 1;40(6):441-445. doi: 10.2500/aap.2019.40.4267. DOI: 10.2500/aap.2019.40.4267
6. Pagnier A, Dermesropian A, Kevorkian-Verguet C, Bourgoin-Heck M, Hoarau C, Reumaux H, Nugues F, Audouin-Pajot C, Blanc S, Carbasse A, Jurquet AL, Voidey M, Villedieu M, Bouillet L, Boccon-Gibod I. Hereditary angioedema in children: Review and practical perspective for clinical management. Pediatr Allergy Immunol. 2024 Dec;35(12):e14268. doi: 10.1111/pai.14268. PMID: 39655944; PMCID: PMC11629734.
7. Lumry WR, Settipane RA. Hereditary angioedema: Epidemiology and burden of disease. Allergy Asthma Proc. 2020 Nov 1;41(Suppl 1):S08-S13. doi: 10.2500/aap.2020.41.200050. PMID: 33109318.
8. Pappalardo E, Cicardi M, Duponchel C, Carugati A, Choquet S, Agostoni A, Tosi M. Frequent de novo mutations and exon deletions in the C1inhibitor gene of patients with angioedema. J Allergy Clin Immunol. 2000 Dec;106(6):1147-54. doi: 10.1067/mai.2000.110471. PMID: 11112899.
9. Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, Aygoeren-Pürsün E, Craig T, Binkley K, Hebert J, Ritchie B, Bouillet L, Betschel S, Cogar D, Dean J, Devaraj R, Hamed A, Kamra P, Keith PK, Lacuesta G, Leith E, Lyons H, Mace S, Mako B, Neurath D, Poon MC, Rivard GE, Schellenberg R, Rowan D, Rowe A, Stark D, Sur S, Tsai E, Warrington R, Waserman S, Ameratunga R, Bernstein J, Björkander J, Brosz K, Brosz J, Bygum A, Caballero T, Frank M, Fust G, Harmat G, Kanani A, Kreuz W, Levi M, Li H, Martinez-Saguer I, Moldovan D, Nagy I, Nielsen EW, Nordenfelt P, Reshef A, Rusicke E, Smith-Foltz S, Späth P, Varga L, Xiang ZY. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010 Jul 28;6(1):24. doi: 10.1186/1710-1492-6-24. PMID: 20667127; PMCID: PMC2921362.
10. Zingale LC, Beltrami L, Zanichelli A, Maggioni L, Pappalardo E, Cicardi B, Cicardi M. Angioedema without urticaria: a large clinical investigation. CMAJ. 2006 Oct 24;175(9):1065-70. doi: 10.1503/cmaj.060535. PMID: 17060655; PMCID: PMC1609157.
11. Gower RG, Busse PJ, Aygören-Pürsün E, Barakat AJ, Caballero T, Davis-Lorton M, Farkas H, Hurewitz DS, Jacobs JS, Johnston DT, Lumry W, Maurer M. Hereditary angioedema caused by c1-esterase inhibitor deficiency: a literature-based analysis and clinical commentary on prophylaxis treatment strategies. World Allergy Organ J. 2011 Feb;4(2 Suppl):S9-S21. doi: 10.1097/WOX.0b013e31821359a2. PMID: 23283143; PMCID: PMC3666183.
12. Bygum A, Aygören-Pürsün E, Beusterien K, Hautamaki E, Sisic Z, Wait S, Boysen HB, Caballero T. Burden of Illness in Hereditary Angioedema: A Conceptual Model. Acta Derm Venereol. 2015 Jul;95(6):706-10. doi: 10.2340/00015555-2014. PMID: 25394853.
C-ANPROM/IT/TAKH/0066